The two ``killer apps'' of modern string processing algorithms have been computational biology and searching the Internet.
While exact matching algorithms are more important in text processing than biology, they are important (1) to illustrate techniques, and (2) as a component of heuristics for approximate matching.
Three primary classes of string matching problems arise, determining whether preprocessing techniques are appropriate:
Suffix trees/arrays are data structures to efficiently support repeated queries on fixed strings.
Such applications justify preprocessing the set of patterns so as to speed search.
Suffix trees solve this problem in linear time, but with excessive complexity and overhead.
We have seen the naive brute force algorithm searching for pattern ![]() in text
in text ![]() in
in ![]() time, where
time, where ![]() and
and ![]() .
.
The brute force algorithm can be viewed as sliding the pattern across from left to right by one position when we detect a mismatch between the pattern and the text.
But in certain circumstances we can slide the pattern to the right by more than one position:
pattern: ABCDABCE text: ....ABCDABCD....
Since we know the last seven characters of the text must be ![]() ,
we can shift the pattern four positions without missing any matches.
,
we can shift the pattern four positions without missing any matches.
Whenever a character match fails, we can shift the pattern forward
according to the failure function ![]() , which is the length
of the longest prefix of
, which is the length
of the longest prefix of ![]() which is a proper suffix of
which is a proper suffix of ![]()
i: 1 2 3 4 5 6 7 8 9 10
P[i]: a b a b a b a b c a
fail[i]: 0 0 1 2 3 4 5 6 0 1
Given this prefix function we can match efficiently - on a character match we increment the pointers, on a mismatch we slide the pattern one step plus the failure function.
a b b b a b a b a b a c a b a b a b a b c a
a b *
*
*
a b a b a b a *
*
a b a b a b a b c a
Note that we will not match the pattern from the begining, but according to where we are in the text - we never look backwards in the text.
Computing the failure function for the pattern can be done in ![]() preprocessing, and hence does not change the asymptotic complexity.
preprocessing, and hence does not change the asymptotic complexity.
If the pattern rejects near the beginning, we did not waste much time in the search.
If the pattern rejects near the end (e.g. EEEEEEEH in ![]() ,
there is an opportunity to slide
it back many positions.
,
there is an opportunity to slide
it back many positions.
Note that we never move backwards through the text to compare a character again - we slide the pattern forward accordingly.
The linearity of KMP follows from an amortized analysis.
Each time we move forward in the text we add ![]() steps to our account,
while each mismatch costs us one step.
steps to our account,
while each mismatch costs us one step.
Provided the account never goes negative, we only did ![]() work total
since there were a total of
work total
since there were a total of ![]() steps put in the account.
steps put in the account.
Thus if many consecutive mismatches requires the pattern to shift repeatedly, it is only because we have enough previous forward moves to compensate.
There are ``improved'' failure functions which do even cleverer preprocessing to reduce the number of pattern shifts. However, such improvements have little effect in practice and are tricky to program correctly.
An alternate linear algorithm, Boyer-Moore, starts matching from right side of the pattern instead of the left side:
pattern: ABCDABCD
text: ABCDABCE....
In this example, the last character in the window does not occur anywhere in the pattern.
Thus we can shift the pattern ![]() positions after one comparison.
positions after one comparison.
Thus the best case time for Boyer-Moore is sub-linear, i.e. ![]() .
The worst-case is
.
The worst-case is ![]() , where
, where ![]() is the number of times the pattern
occurs in the text.
is the number of times the pattern
occurs in the text.
The algorithm precomputes a table of size ![]() describing how far
to shift for a mismatch on each letter of the alphabet, i.e. depending
upon the position of the rightmost
occurrence of that letter in the pattern.
describing how far
to shift for a mismatch on each letter of the alphabet, i.e. depending
upon the position of the rightmost
occurrence of that letter in the pattern.
Further, since we know that the suffix of the pattern matched up to the point of mismatch, we can precompute a table recording the next place where the suffix occurs in the pattern.
We can use the bigger of the two shifts, since no intermediate position can define a legal match.
Boyer-Moore may be the fastest algorithm for alphanumeric string matching in practice when implemented correctly.
The Rabin-Karp algorithm computes an appropriate hash function on
all ![]() -length strings of the text, and does a brute force comparison
only if the hash value is the same for the text window and the pattern.
-length strings of the text, and does a brute force comparison
only if the hash value is the same for the text window and the pattern.
An appropriate hash function is
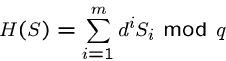
This hash function
can be computed incrementally in constant time
as we slide the window from left to right since
Further, if ![]() is a random prime the expected number of false positives
is small enough to yield a randomized linear algorithm.
is a random prime the expected number of false positives
is small enough to yield a randomized linear algorithm.
Many applications require searching a text for occurrences of any one of many patterns, e.g. searching text for dirty words or searching a genome for any one of a set of known motifs.
Pattern matching with wild card characters (![]() ) is an
important special case of multiple patterns.
) is an
important special case of multiple patterns.
Techniques from automata theory come into play, since any finite
set of patterns can be modeled by regular expressions, and
many interesting infinite sets (e.g. ![]() ) as well.
) as well.
The standard UNIX tool grep stands for ``general regular expression pattern matcher''.
The Aho-Corasick algorithm builds a DFA from the set of patterns and then walks through the text in linear time, taking action when reaching any accepting state.
An important generalization of exact string matching is measuring the distance between two or more strings.
These problems arise naturally in biology because DNA / protein sequences tend to be structurally conserved across species over the course of evolution, so functionally similar genes in different organisms can be detected via approximate string matching.
Computer science applications of approximate string matching included spell checking and file difference testing.
A reasonable distance on strings measure minimizes the cost of the changes which have to be made to convert one string to another. There are three natural types of changes:
Computer scientists usually measure distance between strings ![]() and
and ![]() by the minimum number of insertions, deletions, and substitutions
to transform
by the minimum number of insertions, deletions, and substitutions
to transform ![]() to
to ![]() .
.
Certain mathematical properties are expected of any distance measure, or metric:
Biologists typically instead measure a sequence similarity score which gets larger the more similar the sequences are.
Similar algorithms can be used to optimize both measures.
An elegant algorithm for finding the minimum cost sequence of
changes to transform string ![]() to string
to string ![]() is based on the observation
that the correct action on the rightmost characters of
is based on the observation
that the correct action on the rightmost characters of ![]() and
and ![]() can
be computed knowing the costs of matching various prefixes:
can
be computed knowing the costs of matching various prefixes:
#define MATCH 0 /* symbol for match */
#define INSERT 1 /* symbol for insert */
#define DELETE 2 /* symbol for delete */
int string_compare(char *s, char *t, int i, int j)
{
int k; /* counter */
int opt[3]; /* cost of the three options */
int lowest_cost; /* lowest cost */
if (i == 0) return(j * indel(' '));
if (j == 0) return(i * indel(' '));
opt[MATCH] = string_compare(s,t,i-1,j-1) + match(s[i],t[j]);
opt[INSERT] = string_compare(s,t,i,j-1) + indel(t[j]);
opt[DELETE] = string_compare(s,t,i-1,j) + indel(s[i]);
lowest_cost = opt[MATCH];
for (k=INSERT; k<=DELETE; k++)
if (opt[k] < lowest_cost) lowest_cost = opt[k];
return( lowest_cost );
}
How much time does this program take? Exponential in the length of the strings!
Making this tractible requires realizing that we are repeatedly performing the same computations on each pair of prefixes.
The entire state of the recursive call is governed by the index positions into
the strings.
Thus there are only
![]() different calls.
different calls.
By storing the answers in a table and looking them up instead of recomputing, the algorithm takes quadratic time.
Dynamic programming is the algorithmic technique of efficiently computing recurrence relations by storing partial results. It is very powerful on any ordered structures, like character strings, permutations, and rooted trees.
The table data structure keeps track of the cost of reaching this position plus the last move which took us to this cell.
typedef struct {
int cost; /* cost of reaching this cell */
int parent; /* parent cell */
} cell;
cell m[MAXLEN][MAXLEN]; /* dynamic programming table */
Note how we use and update the table of partial results.
int string_compare(char *s, char *t)
{
int i,j,k; /* counters */
int opt[3]; /* cost of the three options */
for (i=0; i<MAXLEN; i++) {
row_init(i);
column_init(i);
}
for (i=1; i<strlen(s); i++)
for (j=1; j<strlen(t); j++) {
opt[MATCH] = m[i-1][j-1].cost + match(s[i],t[j]);
opt[INSERT] = m[i][j-1].cost + indel(t[j]);
opt[DELETE] = m[i-1][j].cost + indel(s[i]);
m[i][j].cost = opt[1];
m[i][j].parent = 1;
for (k=2; k<=3; k++)
if (opt[k] < m[i][j].cost) {
m[i][j].cost = opt[k];
m[i][j].parent = k;
}
}
goal_cell(s,t,&i,&j);
return( m[i][j].cost );
}
To determine the value of cell ![]() , we need the
the cells
, we need the
the cells ![]() ,
, ![]() , and
, and ![]() .
Any evaluation order with this property
will do, including the row-major order we used.
.
Any evaluation order with this property
will do, including the row-major order we used.
The function string_compare is very general, and must be customized to a particular application.
It uses problem-specific subroutines match and indel to return the costs of character pair transitions:
row_init(int i)
{
m[0][i].cost = i;
if (i>0)
m[0][i].parent=INSERT;
else
m[0][i].parent = -1;
}
|
column_init(int i)
{
m[i][0].cost = i;
if (i>0)
m[i][0].parent=DELETE;
else
m[0][i].parent = -1;
}
|
The functions row_init and column_init to initialize the boundary conditions.
int match(char c, char d)
{
if (c == d) return(0);
else return(1);
}
|
int indel(char c)
{
return(1);
}
|
The function goal_cell returns the desired final cell of interest in the matrix.
goal_cell(char *s, char *t, int *i, int *j)
{
*i = strlen(s) - 1;
*j = strlen(t) - 1;
}
Changing these functions lets us do substring matching, longest common subsequence, and maximum monotone subsequence as special cases.
The cost matrix in converting thou shalt not to you should not:
y o u - s h o u l d - n o t
: 0 1 2 3 4 5 6 7 8 9 10 11 12 13 14
t: 1 1 2 3 4 5 6 7 8 9 10 11 12 13 13
h: 2 2 2 3 4 5 5 6 7 8 9 10 11 12 13
o: 3 3 2 3 4 5 6 5 6 7 8 9 10 11 12
u: 4 4 3 2 3 4 5 6 5 6 7 8 9 10 11
-: 5 5 4 3 2 3 4 5 6 6 7 7 8 9 10
s: 6 6 5 4 3 2 3 4 5 6 7 8 8 9 10
h: 7 7 6 5 4 3 2 3 4 5 6 7 8 9 10
a: 8 8 7 6 5 4 3 3 4 5 6 7 8 9 10
l: 9 9 8 7 6 5 4 4 4 4 5 6 7 8 9
t: 10 10 9 8 7 6 5 5 5 5 5 6 7 8 8
-: 11 11 10 9 8 7 6 6 6 6 6 5 6 7 8
n: 12 12 11 10 9 8 7 7 7 7 7 6 5 6 7
o: 13 13 12 11 10 9 8 7 8 8 8 7 6 5 6
t: 14 14 13 12 11 10 9 8 8 9 9 8 7 6 5
y o u - s h o u l d - n o t
: -1 2 2 2 2 2 2 2 2 2 2 2 2 2 2
t: 3 1 1 1 1 1 1 1 1 1 1 1 1 1 1
h: 3 1 1 1 1 1 1 2 2 2 2 2 2 2 2
o: 3 1 1 1 1 1 1 1 2 2 2 2 2 1 2
u: 3 1 3 1 2 2 2 2 1 2 2 2 2 2 2
-: 3 1 3 3 1 2 2 2 2 1 1 1 2 2 2
s: 3 1 3 3 3 1 2 2 2 2 1 1 1 1 1
h: 3 1 3 3 3 3 1 2 2 2 2 2 2 1 1
a: 3 1 3 3 3 3 3 1 1 1 1 1 1 1 1
l: 3 1 3 3 3 3 3 1 1 1 2 2 2 2 2
t: 3 1 3 3 3 3 3 1 1 1 1 1 1 1 1
-: 3 1 3 3 1 3 3 1 1 1 1 1 2 2 2
n: 3 1 3 3 3 3 3 1 1 1 1 3 1 2 2
o: 3 1 1 3 3 3 3 1 1 1 1 3 3 1 2
t: 3 1 3 3 3 3 3 3 1 1 1 3 3 3 1
Once we have the dynamic programming matrix, we have to walk backwards through it to reconstruct the alignment.
Either explicit back pointers can be kept, or we can remake decisions of how we got to the critical cells starting from the back.
reconstruct_path(char *s, char *t, int i, int j)
{
if (m[i][j].parent == -1) return;
if (m[i][j].parent == MATCH) {
reconstruct_path(s,t,i-1,j-1);
match_out(s, t, i, j);
return;
}
if (m[i][j].parent == INSERT) {
reconstruct_path(s,t,i,j-1);
insert_out(t,j);
return;
}
if (m[i][j].parent == DELETE) {
reconstruct_path(s,t,i-1,j);
delete_out(s,i);
return;
}
}
The actions we take on traceback are governed by match_out, insert_out, and delete_out:
insert_out(char *t, int j)
{
printf("I");
}
delete_out(char *s, int i)
{
printf("D");
}
|
match_out(char *s,char *t,int i,int j)
{
if (s[i] == t[j]) printf("M");
else printf("S");
}
|
The edit sequence from ``thou-shalt-not'' to ``you-should-not'' is DSMMMMMISMSMMMM - meaning delete the first `t', replace the `h' with `y', match the next five characters before inserting an `o', replace `a' with 'u', replace the `t' with a `d'.
By changing the relative costs for matching and substitution we can get different alignments.
If we make the cost of substitution so high as to be greater than the cost of inserting and deleting characters, the returned alignment will seek only to match the largest number of possible characters, returning the longest common subsequence.
int match(char c, char d)
{
if (c == d) return(0);
else return(MAXLEN);
}
This shows us that the exact replacement costs can have a big impact on the optimal alignment.
By changing our traceback functions we get different behavior:
insert_out(char *t,int j)
{
}
delete_out(char *s,int i)
{
}
|
match_out(char *s,char *t,
int i,int j)
{
if (s[i]==t[j])
printf("%c",s[i]);
}
|
abracadabra abbababa length of longest common subsequence = 6 abaaba
Suppose we want to search for a short pattern (say `Skiena' allowing mispellings) in a long text.
Using the approximate string matching function will achieve little sensitivity, since most of the edit cost will be deleting the body of the full text.
We can use the same basic edit distance function for this task, but must adjust the initialization so that the substring matches in the middle of the text are not discouraged:
row_init(int i) /* what is m[0][i]? */
{
m[0][i].cost = 0; /* NOTE CHANGE */
m[0][i].parent = -1; /* NOTE CHANGE */
}
Now the goal cell is the cheapest cell matching the entire pattern:
goal_cell(char *s, char *t, int *i, int *j)
{
int k; /* counter */
*i = strlen(s) - 1;
*j = 0;
for (k=1; k<strlen(t); k++)
if (m[*i][k].cost < m[*i][*j].cost) *j = k;
}
Constructing a meaningful alignment of two sequences requires using a appropriate function to measure the cost of changing between each possible pair symbols.
In correcting text entered by a fast typist, we might penalize pairs of symbols near each other on the keyboard less than those on different sides, for example.
For genomic sequences, the weights/scores governing the cost of changing between bases are given by PAM matrices for ``point accepted mutations''
DNA PAM matrices include Blast similarity and transition / transversion matrices.
A T C G A T C G
A 5 -4 -4 -4 A 0 5 5 1
T -4 5 -4 -4 T 5 0 1 5
C -4 -4 5 -4 C 5 1 0 5
G -4 -4 -4 5 G 1 5 5 0
The four nucleotide bases are classified as either purines (Adenine and Guanine) or pyrimidines (Cytosine and Thymine).
Transitions are mutations which stay within the class (e.g. A![]() G or C
G or C![]() T),
while transversions cross classes (e.g. A
T),
while transversions cross classes (e.g. A![]() C, A
C, A![]() T etc.).
T etc.).
Transitions are more common than transversions.
The following genetic code matrix calculates the minimum number
of DNA base changes to go from a codon for ![]() to a codon for
to a codon for ![]() .
.
Met![]() Tyr requires all 3 positions to change.
Tyr requires all 3 positions to change.
A S G L K V T P E D N I Q R F Y C H M W Z B X A 0 1 1 2 2 1 1 1 1 1 2 2 2 2 2 2 2 2 2 2 2 2 2 S 1 0 1 1 2 2 1 1 2 2 1 1 2 1 1 1 1 2 2 1 2 2 2 G 1 1 0 2 2 1 2 2 1 1 2 2 2 1 2 2 1 2 2 1 2 2 2 L 2 1 2 0 2 1 2 1 2 2 2 1 1 1 1 2 2 1 1 1 2 2 2 K 2 2 2 2 0 2 1 2 1 2 1 1 1 1 2 2 2 2 1 2 1 2 2 V 1 2 1 1 2 0 2 2 1 1 2 1 2 2 1 2 2 2 1 2 2 2 2 T 1 1 2 2 1 2 0 1 2 2 1 1 2 1 2 2 2 2 1 2 2 2 2 P 1 1 2 1 2 2 1 0 2 2 2 2 1 1 2 2 2 1 2 2 2 2 2 E 1 2 1 2 1 1 2 2 0 1 2 2 1 2 2 2 2 2 2 2 1 2 2 D 1 2 1 2 2 1 2 2 1 0 1 2 2 2 2 1 2 1 2 2 2 1 2 N 2 1 2 2 1 2 1 2 2 1 0 1 2 2 2 1 2 1 2 2 2 1 2 I 2 1 2 1 1 1 1 2 2 2 1 0 2 1 1 2 2 2 1 2 2 2 2 Q 2 2 2 1 1 2 2 1 1 2 2 2 0 1 2 2 2 1 2 2 1 2 2 R 2 1 1 1 1 2 1 1 2 2 2 1 1 0 2 2 1 1 1 1 2 2 2 F 2 1 2 1 2 1 2 2 2 2 2 1 2 2 0 1 1 2 2 2 2 2 2 Y 2 1 2 2 2 2 2 2 2 1 1 2 2 2 1 0 1 1 3 2 2 1 2 C 2 1 1 2 2 2 2 2 2 2 2 2 2 1 1 1 0 2 2 1 2 2 2 H 2 2 2 1 2 2 2 1 2 1 1 2 1 1 2 1 2 0 2 2 2 1 2 M 2 2 2 1 1 1 1 2 2 2 2 1 2 1 2 3 2 2 0 2 2 2 2 W 2 1 1 1 2 2 2 2 2 2 2 2 2 1 2 2 1 2 2 0 2 2 2 Z 2 2 2 2 1 2 2 2 1 2 2 2 1 2 2 2 2 2 2 2 1 2 2 B 2 2 2 2 2 2 2 2 2 1 1 2 2 2 2 1 2 1 2 2 2 1 2 X 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2
Amino acids differ to the extent that they like ( hydrophilic) or don't like ( hydrophobic) water.
Hydrophobic residues do not want to be on the surface of a protein.
R K D E B Z S N Q G X T H A C M P V L I Y F W R 10 10 9 9 8 8 6 6 6 5 5 5 5 5 4 3 3 3 3 3 2 1 0 K 10 10 9 9 8 8 6 6 6 5 5 5 5 5 4 3 3 3 3 3 2 1 0 D 9 9 10 10 8 8 7 6 6 6 5 5 5 5 5 4 4 4 3 3 3 2 1 E 9 9 10 10 8 8 7 6 6 6 5 5 5 5 5 4 4 4 3 3 3 2 1 B 8 8 8 8 10 10 8 8 8 8 7 7 7 7 6 6 6 5 5 5 4 4 3 Z 8 8 8 8 10 10 8 8 8 8 7 7 7 7 6 6 6 5 5 5 4 4 3 S 6 6 7 7 8 8 10 10 10 10 9 9 9 9 8 8 7 7 7 7 6 6 4 N 6 6 6 6 8 8 10 10 10 10 9 9 9 9 8 8 8 7 7 7 6 6 4 Q 6 6 6 6 8 8 10 10 10 10 9 9 9 9 8 8 8 7 7 7 6 6 4 G 5 5 6 6 8 8 10 10 10 10 9 9 9 9 8 8 8 8 7 7 6 6 5 X 5 5 5 5 7 7 9 9 9 9 10 10 10 10 9 9 8 8 8 8 7 7 5 T 5 5 5 5 7 7 9 9 9 9 10 10 10 10 9 9 8 8 8 8 7 7 5 H 5 5 5 5 7 7 9 9 9 9 10 10 10 10 9 9 9 8 8 8 7 7 5 A 5 5 5 5 7 7 9 9 9 9 10 10 10 10 9 9 9 8 8 8 7 7 5 C 4 4 5 5 6 6 8 8 8 8 9 9 9 9 10 10 9 9 9 9 8 8 5 M 3 3 4 4 6 6 8 8 8 8 9 9 9 9 10 10 10 10 9 9 8 8 7 P 3 3 4 4 6 6 7 8 8 8 8 8 9 9 9 10 10 10 9 9 9 8 7 V 3 3 4 4 5 5 7 7 7 8 8 8 8 8 9 10 10 10 10 10 9 8 7 L 3 3 3 3 5 5 7 7 7 7 8 8 8 8 9 9 9 10 10 10 9 9 8 I 3 3 3 3 5 5 7 7 7 7 8 8 8 8 9 9 9 10 10 10 9 9 8 Y 2 2 3 3 4 4 6 6 6 6 7 7 7 7 8 8 9 9 9 9 10 10 8 F 1 1 2 2 4 4 6 6 6 6 7 7 7 7 8 8 8 8 9 9 10 10 9 W 0 0 1 1 3 3 4 4 4 5 5 5 5 5 6 7 7 7 8 8 8 9 10
This variety of possible matrix criteria make judging the significance of an alignment tricky.
Most widely used are PAM matrices, for ``point accepted mutations''.
These were constructed by aligning very similar proteins, and tabulating how often each substitution occurred.
The PAM1 matrix scores the transitions when the proteins differ in 1% of the residues.
Raising this to higher powers gives us PAM matrices suited for comparing more distantly related proteins.
Note the main diagonal on these plots of the PAM50 and PAM250 matrices.
More modern than the PAM matrices are the Blosum matrices, reflecting additional sequence alignment data.
The critical problem of biological interest is in comparing two long sequences and finding local areas of similarity.
Typical applications include (1) what regions have been conserved between mouse and human, and (2) recognizing coding regions in `split genes'.
Here using similarity scores is more meaningful than edit distance.
We want ![]() to be the highest-scoring local alignment ending at
to be the highest-scoring local alignment ending at ![]() and
and ![]() .
.
This way to compute this is typically called the Smith-Waterman algorithm.
It is the same basic algorithm as for edit distance, except:
int smith_waterman(char *s, char *t)
{
int i,j,k; /* counters */
int opt[3]; /* cost of the three options */
int match(); int indel();
for (i=0; i<=strlen(s); i++)
for (j=0; j<=strlen(t); j++)
cell_init(i,j);
for (i=1; i<strlen(s); i++)
for (j=1; j<strlen(t); j++) {
opt[MATCH] = m[i-1][j-1].cost + match(s[i],t[j]);
opt[INSERT] = m[i][j-1].cost + indel(t[j]);
opt[DELETE] = m[i-1][j].cost + indel(s[i]);
m[i][j].cost = 0;
m[i][j].parent = -1;
for (k=MATCH; k<=DELETE; k++)
if (opt[k] > m[i][j].cost) {
m[i][j].cost = opt[k];
m[i][j].parent = k;
}
}
goal_cell(s,t,&i,&j);
return( m[i][j].cost );
}
goal_cell(char *s, char *t, int *x, int *y)
{
int i,j; /* counters */
*x = *y = 0;
for (i=0; i<strlen(s); i++)
for (j=0; j<strlen(t); j++)
if (m[i][j].cost > m[*x][*y].cost) {
*x = i;
*y = j;
}
}
int match(char c, char d)
{
if (c == d) return(+5);
else return(-4);
}
int indel(char c)
{
return(-4);
}
cell_init(int i, int j)
{
m[i][j].cost = 0;
m[i][j].parent = -1;
}
Note how the maximum score (31) is not achieved with the exact match public but includes the preceeding blank.
This is because we score a match more than we penalize a mismatch/indel.
b e a t _ r e p u b l i c a n s
: 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0
f: 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0
o: 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0
r: 0 0 0 0 0 0 5 1 0 0 0 0 0 0 0 0 0
_: 0 0 0 0 0 5 1 1 0 0 0 0 0 0 0 0 0
t: 0 0 0 0 5 1 1 0 0 0 0 0 0 0 0 0 0
h: 0 0 0 0 1 1 0 0 0 0 0 0 0 0 0 0 0
e: 0 0 5 1 0 0 0 5 1 0 0 0 0 0 0 0 0
_: 0 0 1 1 0 5 1 1 1 0 0 0 0 0 0 0 0
p: 0 0 0 0 0 1 1 0 6 2 0 0 0 0 0 0 0
u: 0 0 0 0 0 0 0 0 2 11 7 3 0 0 0 0 0
b: 0 5 1 0 0 0 0 0 0 7 16 12 8 4 0 0 0
l: 0 1 1 0 0 0 0 0 0 3 12 21 17 13 9 5 1
i: 0 0 0 0 0 0 0 0 0 0 8 17 26 22 18 14 10
c: 0 0 0 0 0 0 0 0 0 0 4 13 22 31 27 23 19
_: 0 0 0 0 0 5 1 0 0 0 0 9 18 27 27 23 19
g: 0 0 0 0 0 1 1 0 0 0 0 5 14 23 23 23 19
o: 0 0 0 0 0 0 0 0 0 0 0 1 10 19 19 19 19
o: 0 0 0 0 0 0 0 0 0 0 0 0 6 15 15 15 15
d: 0 0 0 0 0 0 0 0 0 0 0 0 2 11 11 11 11
Gaps in sequence alignments occur for several reasons, including (1) the deletion of introns from split genes, (2) the insertion of mobile sequence elements such as transposons, (3) because conserved parts of a protein may include several relatively small docking sites.
Gap in alignments can be modeled as repeated base deletions, where the penalty for a gap is just a linear function of its length.
However, in many applications (e.g. exons/introns, extracting good quotes for the back of a book jacket) the length of the gap is relatively unimportant.
Thus a more meaningful model charges a fixed penalty for the existence of each gap, plus another penalty depending upon the length of the gap.
Affine gap penalties are ![]() for gaps of length
for gaps of length ![]() ,
while logarithmic gap penalties of the form
,
while logarithmic gap penalties of the form ![]() are
suggested by empirical data.
are
suggested by empirical data.
Suppose the gap cost is a completely general function of length for which we cannot assume monotonicity or any other property.
Then we must explicitly try every possible length deletion/insertion at
every possible position, i.e. ![]() takes the best of the following options:
takes the best of the following options:
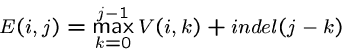
Because we are doing a linear amount of work for each cell, the
time complexity goes to
![]() or
or ![]() if
if ![]() .
.
This algorithm is often called Needleman-Wunsch.
By being clever we can avoid the extra linear cost of
looking for the start of the gap for affine gap penalties,
i.e. penalties of the form ![]() for gaps of length
for gaps of length ![]()
We will use the insertion and deletion recurrences ![]() and
and ![]() to
encode the cost of being in gap mode, meaning we have already paid the
cost of initiating the gap.
to
encode the cost of being in gap mode, meaning we have already paid the
cost of initiating the gap.
With constant amount of work per cell, this algorithm takes ![]() time,
same as without gap costs.
time,
same as without gap costs.
The default in FASTA sets ![]() and
and ![]() , so starting a gap costs
the equivalent of five deletions.
, so starting a gap costs
the equivalent of five deletions.
The special case of convex penalty functions (including logarithmic
costs) can be solved in
![]() time with a more complicated
algorithm.
time with a more complicated
algorithm.
Quadratic space will kill you faster than quadratic time.
![]() bytes equals one gigabyte, while
bytes equals one gigabyte, while ![]() operations
takes 1000 seconds on a machine doing 1 million steps per second.
operations
takes 1000 seconds on a machine doing 1 million steps per second.
The dynamic programming algorithms we have seen only look at neighboring rows/columns to make their decision.
Computing the highest cell score in the matrix does
not require keeping more than then last column and the best value
to date, for a total of ![]() space, where
space, where ![]() .
.
Note that reconstructing the optimal alignment does seem to require keeping the entire matrix, however.
But Hirshberg found a clever way to reconstruct the alignment in ![]() time using only
time using only ![]() space, by recomputing the appropriate portions
of the matrix.
space, by recomputing the appropriate portions
of the matrix.
For each cell, we drag along the row number where the optimal path
to in crossed the middle (![]() nd) column.
nd) column.
This requires only ![]() extra memory, one cell per row.
extra memory, one cell per row.
This works because the crossing point ![]() of the
of the ![]() nd column
means that the optimal alignment lies in the sub matrices
nd column
means that the optimal alignment lies in the sub matrices ![]() from (1,1) to
from (1,1) to
![]() , and
, and ![]() from
from ![]() to
to ![]() .
.
Note that the number of cells in ![]() and
and ![]() totals only half of the the
original
totals only half of the the
original ![]() cells.
cells.
Further, these dynamic programming algorithms are linear in the number of cells they compute.
Thus the total amount of recomputation done is
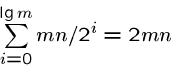
Dynamic programming methods for sequence alignment give the highest quality results.
However, quadratic ![]() algorithms are only feasible for comparing
two modest sized sequences.
algorithms are only feasible for comparing
two modest sized sequences.
Comparing the human genome against mouse with a quadratic algorithm
(
![]() operations)
at a billion operations per second equals 285 years!
operations)
at a billion operations per second equals 285 years!
Comparing your target sequence against the entire database is hopeless, even with special purpose hardware.
Instead, heuristic algorithms such as BLAST and FASTA are used to make an initial scan of the database to find a small number of hits.
Smith-Waterman can then be used to find the optimal alignment to display.
FASTA is a heuristic string alignment program which is based upon
finding short exact matches (![]() -mers) between the query sequence
and the database.
-mers) between the query sequence
and the database.
The trick is to choose ![]() large enough that there are relatively
few hits, but small enough that we are likely to have an exact
large enough that there are relatively
few hits, but small enough that we are likely to have an exact
![]() -mer match between related sequences.
-mer match between related sequences.
Recommended values of ![]() are 2 for protein sequences and 6 for
DNA sequences.
are 2 for protein sequences and 6 for
DNA sequences.
Note that there are at most ![]() distinct
distinct ![]() -mers in a sequence of
length
-mers in a sequence of
length ![]() .
.
A hash table of all ![]() -mers in the database can be built once
and used repeatedly for efficiently looking up the
-mers in the database can be built once
and used repeatedly for efficiently looking up the ![]() -mers
in query strings.
-mers
in query strings.
A dynamic programming-like algorithm is used to align the ![]() -mer
hits between query and database, but the problem is much smaller
since there are far fewer hits than bases.
-mer
hits between query and database, but the problem is much smaller
since there are far fewer hits than bases.
BLAST stands for ``Basic Local Alignment Search Tool''.
It also seeks to exploit the speed of exact pattern matching while factoring in the impact of the scoring matrix.
BLAST also breaks the query into ![]() -mers in order to search
a hash table, but for input
-mers in order to search
a hash table, but for input ![]() -mer
-mer ![]() constructs all other
constructs all other
![]() -mers which lie within a distance
-mers which lie within a distance ![]() of
of ![]() .
.
By processing all these patterns into an automata, BLAST can then make one linear-time pass through the database to find all exact matches and group them in an alignment.
The window size ![]() is typically 3-5 for protein sequences and 12 for DNA
sequences.
is typically 3-5 for protein sequences and 12 for DNA
sequences.
Conventional wisdom has BLAST as faster than FASTA, but perhaps a little less accurate.
Several variants of the Basic Local Alignment Search Tool are available at www.ncbi.nlm.nih.gov/BLAST/
You can download your own local copy of the code and databases, or use web resources.
Database choices include:
Your query sequence can be (1) an amino acid or nucleotide sequence you type or paste in, or (2) the accession or GI number of Genbank entry.
There are a wide range of output formats.
You can select the organism you are interested in to limit your search.
You can change the expect value ![]() , the threshold for reporting matches against a database sequence.
The default threshold of 10 means that 10 matches are expected to be found merely by chance (Karlin and Altschul).
Lower expect thresholds are more stringent, leading to fewer chance matches being reported.
, the threshold for reporting matches against a database sequence.
The default threshold of 10 means that 10 matches are expected to be found merely by chance (Karlin and Altschul).
Lower expect thresholds are more stringent, leading to fewer chance matches being reported.
The expect value decreases exponentially with the alignment score ![]() .
.
You can use filter to masks sequences of low compositional complexity, i.e. eliminate statistically significant but biologically uninteresting reports.
You can select the cost comparison matrix. The default is BLOSUM62, but with this matrix fairly long alignments are requires to rise above background. PAM matrices are recommends if search for short alignments.
To assess whether a given alignment constitutes evidence for homology, it helps to know how strong an alignment can be expected from chance alone.
A model for expected number of high-scoring segment pairs (HSPs) with score at least ![]() is
is
Doubling length of either sequence doubles ![]() , as it should.
, as it should.
Doubling the score for an HSP to ![]() requires it to attain the score x twice in a row,
so
requires it to attain the score x twice in a row,
so ![]() should decrease exponentially with score.
should decrease exponentially with score.
The number of random HSPs with score ![]() is described by a Poisson
distribution, so the probability of finding exactly
is described by a Poisson
distribution, so the probability of finding exactly ![]() HSPs with
score
HSPs with
score ![]() is given by
is given by
![]() .
.
Hence the probability of finding 0 HSPs is ![]() , and the probability of
finding at least one is
, and the probability of
finding at least one is ![]() .
This is the p-value of the score.
.
This is the p-value of the score.
A statistical method to measure significance is to generate many random sequence pairs of the appropriate length and composition, and calculate the optimal alignment score for each.